 |
| Barbie and Ken |
That is what the Ken-Cat has done with David Burges, the science editor of the Testimony magazine, in this recent article on his Evolutionary Creationism blog. In a Universe far, far away, the gluons and quarks of David Burges are probably even now reforming themselves to create a new Christadelphian Young Earth Creationist who will one day argue that his God magically created that Universe six thousand years before his birthday.
 |
| The current Testimony magazine committee. David Burges is upper left; Bernard Burt is on the far right. |
In another time and another place a Ken-Cat think-alike must be sharpening his claws and raising the fur on the back of his neck to send the David Burges clone to meet his maker for a second time. :)
But enough of my mad philosophising. I give to you my dear readers what surely has to be the ultimate, the decisive, the conclusive Ken-Cat Anti-Young Earth Creationist article. Opposition to this sort of scholarship, passion and determination must be impossible. YEC resistance is futile and unthinkable. They have to hand over the keys to their castle of self-delusion and accept that they can no longer take the awesome bombardment of their flattened walls of fideistic defence.
 |
| The Ken-Cat strikes again |
By Christadelphian Ken Gilmore
How does Burges attempt to deal with the overwhelming evidence from comparative anatomy, developmental biology, comparative genomics, biogeography, and palaeontology that confirms the reality of common descent? In short, he ignores it, or tries to peddle the long-rebutted "common design" argument. Given the amount of shared design flaws from the gross anatomical to the genomic, this 'common design argument' readily is turned back on those who peddle it as it demonstrates that the intelligent designer not only made mistakes, but made exactly the same mistakes repeatedly, and did so in exactly the right way to simulate common descent. This makes any intelligent designer incompetent and deceptive.
Worse still is when Burges attempts to blame some of the genomic errors on the Fall, which immediately raises the question of why God cursed humans, apes, and monkeys with exactly the right pattern of pseudogenes, retrotransposons, and ERV elements as to simulate common descent. Finally, Burges even resorts to the "God buried the fossils" school of deception by stating that "…these features of DNA look like common descent, and therefore must evidence it, because otherwise God would be deceiving us. But while He is certainly "the God of truth"… it is also true that, for those who have turned from Him, He can send strong delusion that they should believe a lie…It is those who absolutely trust His Word who will not be deceived." [1]
This last argument not only is grossly offensive as it asserts without any evidence that any intellectually honest believer who accepts that the evidence for common descent is beyond reasonable doubt, and recognises the utterly threadbare arguments offered by the special creationists has automatically turned from God. That alone is an outrageous slur on Burges' fellow believers.
Furthermore, it betrays theological incompetence as the 2 Thessalonians 2:11 reference has absolutely nothing to do with evolutionary biology, and is simply an example of tearing a verse completely out of context in order to provide a semblance of divine approval to his science denialism.
It is also an admission of defeat as it concedes that the evidence for common descent is overwhelming, but seeks to nullify the power of that evidence by assuming without any evidence that God has deliberately written a superfluous lie into creation across multiple areas, ranging from a faked biogeographical distribution of species, through to a faked fossil record complete with innumerable transitional fossils, to a faked genome complete with multiple shared genomic glitches; not just retrotransposons, ERVs, and pseudogenes, but shared faked evidence of DNA repair - non-homologous end joining. This scale of deception would make YHWH out to be a monstrous deceiver.
Finally, Burges endorses fideism by declaring that "it is those who absolutely trust His Word who will not be deceived" which is simply a fundamentalist Christadelphian analog of the AiG statement of faith that declares:
By definition, no apparent, perceived or claimed evidence in any field, including history and chronology, can be valid if it contradicts the scriptural record. Of primary importance is the fact that evidence is always subject to interpretation by fallible people who do not possess all information.
It is effectively closing ones eyes and ears to the problems and loudly screaming away all evidence to the contrary, and shows just how far our community has degenerated since its early days where our pioneers were honest enough to engage the evidence on its merits, rather than hide from it in terror. Of course, what Burges and his fellow science denialists forget is that Biblical evidence is always subject to interpretation by fallible people. Egregious as his scientific blunders are, it is his flawed exegesis that is ultimately the more worrying problem.
Genetic plagiarism - genomic confirmation of the fact of common descent
In Michael Behe's widely panned book The Edge of Evolution (to which Burges vainly appealed in an attempt to bolster his assertion that Darwinian evolution was mathematically impossible), Behe neatly summarised why shared identical genomic 'glitches' are regarded as compelling evidence for common descent:
When two lineages share what appears to be an arbitrary genetic accident, the case for common descent becomes compelling, just as the case for plagiarism becomes overpowering when one writer makes the same unusual misspellings of another, within a copy of the same words. That sort of evidence is seen in the genomes of humans and chimpanzees. For examples, both humans and chimps have a broken copy of a gene that in other mammals helps make vitamin C As a result, neither humans nor chimps can make their own vitamin C.
[…]
The same mistakes in the same gene in the same positions of both human and chimp DNA. f a common ancestor first sustained the mutational mistakes band subsequently gave rise to these two modern species, that would very readily account for both why both species have them how. It's hard to imagine how there could be stronger evidence for common ancestry of chimps and humans. [2]
The logic is clear, and unarguable. When we examine the genomes of humans and other animals, we find plenty of examples of shared genetic 'mistakes' at exactly the same place in their genomes. This is correctly regarded as overwhelming evidence for common ancestry, with the original genetic error occurring in a species ancestral to the currently living ones, and subsequently being inherited.
The term 'genetic error' is of course a non-technical term employed to get across the essential idea.These 'errors' refer to:
- pseudogenes - genetic elements resembling genes but which are non-functional due to genetic damage
- retrotransposons - mobile genetic elements which copy and paste themselves randomly throughout the genome
- endogenous retroviral elements - decayed remnants of retroviral infection which integrated into the germ line and became inherited along with the rest of the genome
- non-homologous end-joining (NHEJ) - evidence of DNA repair where external genomic elements have been imported to repair a defect
- nuclear sequences of mitochondrial origins (NUMTS) - pieces of mitochondrial DNA that have been transposed into nuclear DNA
- interstitial telomeric sequences - elements of telomeric DNA (normally found at the ends of chromosomes) found within chromosomes
The presence of any of these shared identical genomic elements at the same place in related species is as Behe acknowledged, prima facie evidence of common descent.
Pseudogenes
A pseudogene is a genetic element closely resembling a gene, but which in general is not able to code for its intended product. There are three classes of pseudogenes: unitary, duplicate and processed.
1. Unitary pseudogenes occur when a gene suffers a crippling mutation which renders it unable to function. The classic example of a unitary pseudogene is the GULO pseudogene. In most animals, GULO codes for the enzyme L-gulono-γ-lactone oxidase, the terminal enzyme in the biosynthesis of ascorbic acid, or vitamin C. In humans, apes, monkeys, guinea pigs and a few other animals, the GULO gene is a pseudogene, having been crippled by a lethal mutation, which means they are unable to synthesise vitamin C, and have to rely on dietary ascorbic acid.
2. Duplicated pseudogenes occur when a functional gene is copied and picks up mutations which result in it becoming non-functional. As the organism already has a functional copy of the first gene, the presence of a duplicate gene which has lost function through mutation will not affect the organism in any substantive manner. Examples of duplicated pseudogenes include the ψη-globin pseudogene and the CYP21 pseudogene. The former is a haemoglobin pseudogene, the latter is a pseudogene version of the gene coding for cytochrome P450 C21 which when functional is involved in steroid biosynthesis.
3. Processed pseudogenes occur when an RNA transcript of a gene is reverse transcribed randomly back into the genome. Normally, after DNA is transcribed to RNA, the introns (long non-coding sections in the gene) are removed, and a section of RNA called a poly A tail (used in assisting the transport of the RNA out of the nucleus and in assisting the translation process, where the RNA is used as the template for protein synthesis) is added. Transcription normally results in the creation of an RNA copy of a DNA gene, but the phenomenon of reverse transcription will create a DNA copy of RNA. Reverse transcription will copy the processed RNA transcript back into the genome at a random location. These are easily recognised as processed pseudogenes as they lack the introns of the normal gene, and possess the poly A tail which is not present in the DNA original. As they lack the promoter sequences (regulatory sequences near the gene which are critical to initiate transcription.

Formation of processed and duplicated pseudogenes. Source
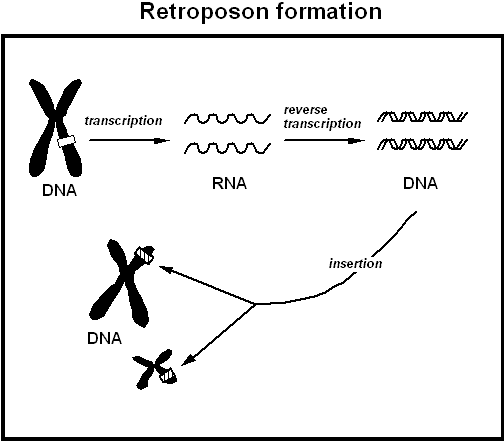
Retrotransposons
Retrotransposons are mobile genetic elements which replicate by the creation of an RNA transcript of themselves which is reverse transcribed to a DNA copy which is randomly inserted back into the genome. This process has allowed them to amplify their number to such a degree that approximately 40% of the human genome consists of multiple copies of these genetic parasites.

Source
{kind=link}
There are two classes: LTR retrotransposons and non-LTR retrotransposons. The former are related to retroviruses, but unlike them have a completely intracellular life. They will not be considered further. Non-LTR retrotransposons include LINEs and SINEs.
LINEs - Long Interspersed Nuclear Elements are genetic elements around 7000 base pairs long which have the code for the reverse transcriptase enzyme. In theory, they are able to reverse transcribe their own RNA copies into DNA and insert this copy randomly back into the genome. Some LINEs are mutated, so they are unable to continue the retrotransposition cycle, while others are still functional. They make up around 20% of the genome
SINEs - Short Interspersed Nuclear Elements are short genetic elements (around 500 base pairs long) which do not have their own copy of reverse transcriptase; they are reliant on other transposable elements to aid in their transposition. Around 13% of the human genome is made up of SINEs
Retrotransposable elements are genetic parasites - they copy and paste themselves randomly throughout the genome. Uncommonly, the genome co-opts retrotransposable elements and creates a genomic element with a new function. Generally, retrotransposons are classic junk DNA, providing no benefit to the host genome and at times being implicated in genetic disease. [3-6] Needless to say the fact that nearly half of our genome is composed of parasitic DNA which can cause disease is impossible to honestly reconcile with intelligent design, but makes perfect sense under an evolutionary model where these elements copy and paste themselves randomly, causing the genome size to grow over time and contribute to genomic instability and disease.
Endogenous Retroviruses
Retroviruses are RNA viruses that reproduce intracellularly by using their reverse transcriptase enzyme to produce a DNA copy of their genome which is then inserted into the host genome. Once the DNA copy is part of the host cell, the host cell genetic replication machinery produces new copies of the retrovirus.

Retroviral life cycle. Source
{kind=link}
If the retrovirus integrates into the host's germ line, then it can be passed down to the next generation. When that happens, it becomes an endogenous retrovirus. As the DNA copy does not produce material essential to the well-being of the cell (as one would expect given its viral origins) it will eventually become inactivated by mutation. The presence of endogenous viral elements in an organism's genome is proof of a prior retroviral infection. When two related organisms share the same ERV at exactly the same place in the genome, we have powerful evidence that these organisms share a common ancestor in which the original viral infection took place and was then passsed down to the descendant species.
Special creationists are fond of comparing the human genome to an encyclopaedia, but the truth is that if your genome was represented by a 100 volume encyclopaedia, most of it would be gibberish, with only around 20-30 at most containing meaningful information:

Source. Image from The Genome by Numbers, the Welcome Trust.
Over half of our genome is parasitic DNA such as LINEs, SINEs, ERVs and pseudogenes. Just the existence of this almost completely non-functional parasitic material is impossible to square with an intelligent design of the genome, but makes sense only in the light of an evolutionary origin of the genome.
NHEJ
NHEJ is one of the main mechanisms [7] used to repair double stranded DNA breaks. It can be used to repair DNA breaks which normally occur such as in immunoglobulin and T cell receptor recombination, as well as damage to DNA caused by environmental damage.
There are four steps involved in NHEJ: detecting the DNA break, bridging both DNA ends, preparing the ends to make them suitable for joining, and the final repair. As a direct consequence of the repair method, NHEJ leaves a 'scar.'
 |
| Outline of NHEJ. Source |
NUMTS
NUMTS occur when an element of mtDNA becomes incorporated into nuclear DNA. They range in size from 31 bases to almost the entire length of the mitochondrial genome. Sometimes, mtDNA elements have been used to effect repairs in nuclear DNA. Needless to say, shared identical sections of repaired DNA using mtDNA would be hard to ascribe to design.
Interstitial telomeric sequences
Telomeric DNA is found at the end of chromosomes, and exist to prevent the ends of chromosomes being mistaken for DNA breaks and being joined together. They have a distinctive structure consisting of multiple repeats of the sequence TTAGG. When we find the characteristic signature of telomeric DNA within chromosomes, we have evidence either of chromosomal fusion, or insertion via the action of telomerase. |
| One way in which NUMTS are formed. Source |
 |
| Telomere location. Source |
{kind=link}
Shared pseudogenes
The enzyme cytochrome P450 C21 is of critical importance in the biosynthesis of steroid hormones. Humans have both a working copy of the CYP21 gene, as well as a pseudogene, which is damaged in three main ways:
- An eight base pair deletion in exon 3 of the gene
- A one base pair substitution at codon 318 of exon 8
- A single nucleotide insertion in exon 7
Kawaguchi et al analysed the DNA of humans, orangutans, chimpanzees and gorillas to clarify how and when the defects in the CYP21 pseudogene occurred:
The primary purpose of this study has been to determine the evolutionary origins of the three defects characterizing the human CYP21P gene. The study shows that the 8-bp deletion in exon 3 is present in the chimpanzee but not in the gorilla or orangutan genes, whereas the T insertion in exon 7 and the substitution generating the stop codon in exon 8 are restricted to human genes. [8]
In other words, the 8 base pair deletion occurred in a common ancestor of humans and chimpanzees, while the substitution and insertion occurred after the human-chumpanzee speciation event:
Our results are consistent with this scenario: the 8-bp deletion apparently occurred after the gorilla lineage split off but before the chimpanzee and human lineages separated from each other. We can thus date the occurrence of the 8-bp deletion rather precisely within a relatively short period of some 6 Myr ago. The deletion was followed, in the human lineage, by the two other defective mutations. [9]
When we have identical errors in the CYP21 pseudogene of chimpanzees and humans, it stretches credibility to assume that:
- Purely by chance, chimpanzees and humans both have a CYP21 pseudogene which arose via a duplication event
- Purely by chance, they both acquired the same eight base pair deletion
Other examples of shared pseudogenes follow:
1. The GBA gene, which codes for the enzyme glucocerebrosidase has a pseudogene present not only in humans, but in chimpanzees and gorillas. Human, chimpanzees and gorillas share the same 55 base pair deletion. [10]
2. The RT6 gene normally codes for a protein which is found on the surface membrane of T lymphocytes (a class of white blood cell). In both humans and chimpanzees it is a pseudogene, which means this protein is not expressed. Haag et al have examined the human and chimpanzee RT6 pseudogene:
3. Humans and apes are unable to synthesise the enzyme urate oxidase, as the gene which normally would code for it is a pseudogene. In humans, chimpanzees, this is due to the same mutation. In gibbons, which are also unable to synthesise urate oxidase, this is due to a separate mutation event:
1. The GBA gene, which codes for the enzyme glucocerebrosidase has a pseudogene present not only in humans, but in chimpanzees and gorillas. Human, chimpanzees and gorillas share the same 55 base pair deletion. [10]
2. The RT6 gene normally codes for a protein which is found on the surface membrane of T lymphocytes (a class of white blood cell). In both humans and chimpanzees it is a pseudogene, which means this protein is not expressed. Haag et al have examined the human and chimpanzee RT6 pseudogene:
We have now cloned and sequenced the homologues of the RT6 genes from humans of distinct ethnic backgrounds and of the chimpanzee. Surprisingly, in each case, three premature in-frame stop codons preclude expression of the single copy RT6 gene as a cell surface protein. Otherwise, the RT6 genes of human and chimpanzee exhibit high structural conservation to their rodent counterparts. RNA expression analyses indicate that the RT6 gene is not transcriptionally active in human T cells or any other human tissue analyzed so far. To our knowledge, RT6 represents the first mammalian membrane protein identified that has been lost universally in the human and chimpanzee species due to gene inactivation. [11]Again, here is an example of humans and chimpanzees having the same pseudogene with the same crippling mutation, which makes sense if humans and chimpanzees shared a common ancestor in which this mutation took place and from whom the two lines inherited the pseudogene. From a special creationist point of view, this is inexplicable.
3. Humans and apes are unable to synthesise the enzyme urate oxidase, as the gene which normally would code for it is a pseudogene. In humans, chimpanzees, this is due to the same mutation. In gibbons, which are also unable to synthesise urate oxidase, this is due to a separate mutation event:
Two nonsense mutations at codon positions 33 and 187 and an aberrant splice site were found in the human gene. These three deleterious mutations were also identified in the chimpanzee. The nonsense mutation at codon 33 was observed in the orangutan urate oxidase gene. None of the three mutations was present in the gibbon; in contrast, a 13-bp deletion was identified that disrupted the gibbon urate oxidase reading frame. These results suggest that the loss of urate oxidase during the evolution of hominoids could be caused by two independent events after the divergence of the gibbon lineage; the nonsense mutation at codon position 33 resulted in the loss of urate oxidase activity in the human, chimpanzee, and orangutan, whereas the 13-bp deletion was responsible for the urate oxidase deficiency in the gibbon. [12]
4. Humans have thirteen genes which code for the oxygen-carrying molecule haemoglobin. Four of them in adults are functional, while five are active only in the foetal stage and are inactivated around birth. The remaining four are pseudogenes. The genes occur in two clusters, the alpha cluster (three pseudogenes and four genes) and the beta cluster (one pseudogene and five genes).
The single pseudogene in the beta cluster - the ψβ pseudogene - is considerably degraded, with around 30% of the gene sequence mutated when compared with the functional β haemoglobin gene. Given the known rate of mutation, this means the ψβ pseudogene must have been disabled a long time ago. Therefore, common descent would predict we'd see it not only in humans and the great apes, but in more distantly related primates, and that is exactly what we see. The ψβ pseudogene is seen in humans, apes, baboons and new world monkeys. Furthermore, when we examine the ψβ pseudogene in humans, chimpanzees and gorillas, we see the same crippling mutations:
These three pseudogenes each share the same substitutions in the initiator codon (ATG → GTA), a substitution in codon 15 which generates a termination signal TGG → TGA, nucleotide deletion in codon 20 and the resulting frame shift which yields many termination signals in exons 2 and 3. [13]One of the pseudogenes in the alpha cluster, the ψζ pseudogene is almost identical with the working ζ foetal haemoglobin gene. It is another example of a duplicated pseudogene, one which was inactivated only a relatively short time ago [14]. When we look at the genome of the chimpanzee, regarded as the closest living relative of humans, we see two ζ haemoglobin genes, confirming that the conversion of the duplicated ζ gene to a pseudogene took place after the human-chimp speciation event, relatively recently.

Source: Fairbanks D.J. "Relics of Eden: The Powerful Evidence of Evolution in Human DNA"
5. Finally, humans, apes and monkeys, unlike most other animals, are unable to synthesise their own vitamin C. As one would have guessed by now, this is because one of the enzymes involved in the biosynthesis of vitamin C, L-gulonolactone oxidase is not synthesised as GULO, the gene that codes for the enzyme, is a pseudogene in humans, apes and monkeys. Furthermore, it is crippled in exactly the same way [15], consistent with the gene being converted to a pseudogene in the common ancestor of humans, apes and monkeys.
Furthermore, when we examine the GULO pseudogene in these primates, we find random mutations, which is what one would expect in a section of DNA that is selectively neutral. Common descent would predict that primates that share a recent common ancestor would differ by fewer random mutations than those that share a remote common ancestor. This is exactly what we see. Furthermore, when we plot this data in tree form, we get an evolutionary family tree consistent with the standard one obtained from morphological data.
Furthermore, when we examine the GULO pseudogene in these primates, we find random mutations, which is what one would expect in a section of DNA that is selectively neutral. Common descent would predict that primates that share a recent common ancestor would differ by fewer random mutations than those that share a remote common ancestor. This is exactly what we see. Furthermore, when we plot this data in tree form, we get an evolutionary family tree consistent with the standard one obtained from morphological data.

{kind=link}
This consonance between morphological and molecular phylogenetic trees is exactly what common descent would predict, and utterly impossible to honestly reconcile with common design, unless God was deliberately creating life with errors in order to fake common descent.
This barely touches the surface of the vast array of pseudogene evidence demonstrating human-ape common ancestry. As molecular biologist Daniel Fairbanks notes:
With a few notable exceptions, chimpanzees and humans have the same pseudogenes in the same places, and they are, on average, about 98 percent similar. [16]Needless to say, the odds of these two species purely by chance having the same genes convert to pseudogenes with exactly the same genetic mutation causing the pseudogenisation of these genes is so remote as to be impossible. Common design fails to explain this evidence:
- It fails to explain why, if a creator did not intend for humans and apes to have these genes in the first place, were they created with broken versions of genes that in other animals are functional.
- It fails to explain why these broken genes were created in closely related animals with the same crippling mutation.
- It fails to explain why these broken genes were also created with random mutations places in exactly the right way to allow scientists to construct evolutionary family trees that are consonant with the standard evolutionary family trees. Such consonance is exactly what one would expect if common descent was true. Special creation is simply unable to honestly account for this evidence.
The evidence for common descent just from pseudogenes is clear, and unarguable. It is however not the only source of evidence for common descent from shared genetic 'errors'.
Shared retrotransposons
Retrotransposons are mobile genetic elements that copy and paste themselves randomly throughout the genome. Essentially, they are genetic parasites. More importantly, their presence is evidence of a prior insertion event in the genome - any argument that they were specially created can be dismissed, if only because of the considerable evidence [17-20] linking retrotransposons with genetic disease.
As the presence of retrotransposons in the genome is evidence of prior copying and pasting of that genetic element, if we see the same retrotransposon in exactly the same place in the genome of two related species, then we have two options:
Shared retrotransposons
Retrotransposons are mobile genetic elements that copy and paste themselves randomly throughout the genome. Essentially, they are genetic parasites. More importantly, their presence is evidence of a prior insertion event in the genome - any argument that they were specially created can be dismissed, if only because of the considerable evidence [17-20] linking retrotransposons with genetic disease.
As the presence of retrotransposons in the genome is evidence of prior copying and pasting of that genetic element, if we see the same retrotransposon in exactly the same place in the genome of two related species, then we have two options:
- Purely by chance, the same retrotransposon inserted itself into exactly the same place in human and ape genomes
- The retrotransposon was first inserted in the genome of the common ancestor of human and ape, and then inherited by the descendant species.
The odds of independent identical retrotransposon insertion at identical places in human and ape genomes is remote. If we have an example where seven retrotransposons are found in exactly the same places in human and chimpanzee genomes, then the chances are so remote as to be impossible. This is exactly what we have found. In the alpha haemoglobin cluster of humans and chimpanzees. Researchers have found that "[i]n each case, with the exception of minor sequence differences, the identical Alu repeat is located at identical sites in the human and chimpanzee genomes." [21]
In fact, by looking for the presence of retrotransposons in primate genomic data, we have been able to construct a reliable evolutionary family tree that is consistent with the accepted tree based on morphological data. [22]
In fact, by looking for the presence of retrotransposons in primate genomic data, we have been able to construct a reliable evolutionary family tree that is consistent with the accepted tree based on morphological data. [22]

Source: Gene (2007) 390:39-51
The use of the unique pattern of retrotransposon data has real-world applications in allowing researchers to identify - for example - what sort of primates a carnivore eats based on the genetic material detected in its faeces. It also allows accurate identification of products seized in the illegal wildlife trade. [23] It is also worth noting that this approach can be used to identify humans. [24] Special creationists who accept the result of a paternity test should, to be consistent, accept this technique when it confirms common descent of humans and apes.
Shared endogenous retroviral elements
The evidence for common descent from retrotransposons and pseudogenes is overwhelming, so looking at what endogenous retroviral elements have to say may appear overkill. However, the ERV data is perhaps the most compelling of all. While pseudogenes and retrotransposons are 'indigenous' to the species in question for want of a better term, ERV data is unarguably alien, presence of an ancient retroviral infection that became integrated into the germ line of the animal, and passed down the generations. Therefore, the presence of identical retroviral elements at the same position in human and ape genomes strongly suggests infection and integration of the retroviral element in a species ancestral to human and ape.
Shared endogenous retroviral elements
The evidence for common descent from retrotransposons and pseudogenes is overwhelming, so looking at what endogenous retroviral elements have to say may appear overkill. However, the ERV data is perhaps the most compelling of all. While pseudogenes and retrotransposons are 'indigenous' to the species in question for want of a better term, ERV data is unarguably alien, presence of an ancient retroviral infection that became integrated into the germ line of the animal, and passed down the generations. Therefore, the presence of identical retroviral elements at the same position in human and ape genomes strongly suggests infection and integration of the retroviral element in a species ancestral to human and ape.
John Coffin, an acknowledged expert in virology notes that:
Because the site of integration in the genome, which comprises some three billion base pairs in humans, is essentially random, the presence of an ancient provirus at exactly the same position in different, but related, species cannot occur by chance, but must be a consequence of integration into the DNA of a common ancestor of all the species that contain it. It evolution of retroviruses follows, therefore, that we can infer what viruses were present millions of years ago by examining the distribution of endogenous proviruses in modern species. [25]
Johnson, in a paper coauthored with Welkin Johnson [26] showed that ERV elements can be used to construct primate evolutionary family trees. This hinges on the principle that ERVs, once integrated into the germ line will be inherited, and therefore of use as markers of inheritance. They used human endogenous retroviruses (HERVs) and found that these HERVs are:
the result of integration events that took place between 5 and 50 million years ago, as indicated by the distribution of specific proviruses at the same integration sites (or loci) among related species. The evolution of primates has been the subject of intense study for well over a century, providing a well established phylogenetic consensus with which to compare and evaluate the performance of ERVs as phylogenetic markers. [27]
The idea behind this is fairly simple. An ERV element should not be under positive selection as it is of no use to the organism, and will eventually accumulate random mutations. The longer the time between the divergence of the two lines leading to the modern speies, the more mutations will accumulate in these ERV elements. From this data, an evolutionary family tree can be constructed.
ERV elements differ from pseudogenes and retrotransposons in that they have three sources of information that allow evolutionary family trees to be constructed:
- The distribution of ERVs among related species
- Accumulated mutations in ERVs, allowing an estimate of genetic distance
- Sequence divergence between the LTRs at each end of the ERV, which is a source of information unique to endogenous retroviruses.
The odds of this distribution of ERV elements occuring by chance is remote. The vertebrate genome is huge, and retroviral integration is random, making the odds of identical ERV integration at the same place in multiple genomes unlikely:
Therefore, an ERV locus shared by two or more species is descended from a single integration event and is proof that the species share a common ancestor into whose germ line the original integration took place. Furthermore, integrated proviruses are extremely stable: there is no mechanism for removing proviruses precisely from the genome, without leaving behind a solo LTR or deleting chromosomal DNA. The distribution of an ERV among related species also reflects the age of the provirus: older loci are found among widely divergent species, whereas younger proviruses are limited to more closely related species. [28]
The second point has been addressed previously, and need not be covered again. The final point is one unique to ERVs. At each end of the ERV is a sequence known as a LTR, or Long Terminal Repeat. The mechanics of reverse transcription mean that both LTRs will be identical when the ERV integrates into the genome. Johnson and Coffin note:
Furthermore, both clusters are predicted to have similar branching patterns as determined by the phylogenetic history of the host species, with similar branch lengths. Thus, each tree displays two estimates of host phylogeny, both of which are derived from the evolution of an initially identical sequence. As we shall see, deviation of actual trees from this prediction provides a powerful means of testing the assumptions and detecting events other than neutral accumulation of mutations in the evolutionary history of a species. [29]
Johnson and Coffin looked at the distribution of ERVs in the primate genetic material analused, and found:
Three of the loci, HERV-KC4, HERV-KHML6.17, and RTVL-Ia, were detectable in the genomes of OWMs and hominoids, but not New World monkeys, and therefore integrated into the germ line of a common ancestor of the Old World lineages. HERV-K18, RTVL-Ha, and RTVL-Hb were found exclusively in humans, gorillas, chimpanzees, and bonobos, and thus are consistent with a gorilla/chimpanzee/human clade. None of the loci was detected in New World monkeys. [30]
This is perfectly explained by common descent. To reiterate an “ERV locus shared by two or more species is descended from a single integration event and is proof that the species share a common ancestor into whose germ line the original integration took place.” Johnson and Coffin found many loci shared by these primate species, some shared only by humans, chimps, bonobos and gorillas, some shared only by old world monkeys and hominoids (humans and great apes). This data is consistent with an evolutionary origin of these species, but impossible to explain by special creation.
Most of the ERVs analysed produced phylogenetic trees consistent with expectation. Their conclusions:
The HERVs analyzed above include six unlinked loci, representing five unrelated HERV sequence families. Except where noted, these sequences gave trees that were consistent with the well established phylogeny of the old world primates, including OWMs, apes, and humans… Phylogenetic analysis using HERV LTR sequences gives rise to trees with a predictable topology, on which is superimposed the phylogeny of the host taxa, and allows ready detection of conversion events. [31]
Other studies show that humans and primates share ERVs in a way consistent with common descent. Barbulescu et al showed that many human ERVs of the HERV-K class (present in humans, apes and old world monkeys) are unique to humans:
Two proviruses, HERV-K105 and HERV-K110/HERV-K18 were detected in both humans and apes. HERV-K110 was present in humans, chimpanzees, bonobos and gorillas but not in the orangutan. Thus, this provirus formed after orangutans diverged from the lineage leading to gorillas, chimpanzees, bonobos and humans, but before the latter species separated from each other. HERV-K105 was detected in humans, chimpanzees and bonobos, but not in gorillas or the orang-utan. The preintegration site, however, could not be detected in gorillas or orang-utans using several different primers based on the human sequences that flank this provirus. It is therefore unclear from this analysis whether this provirus formed after gorillas diverged from the human–chimpanzee–bonobo lineage, or if it formed earlier but was subsequently deleted in one or more lineages leading to modern apes. It is clear that at least one full-length HERV-K provirus in the human genome today has persisted since before humans, chimpanzees, bonobos and gorillas separated during evolution, while at least eight formed after humans diverged from the extant apes. [32]
Belshaw et al, looking at the long-term reinfection of the human genome by ERVs note that:
Within humans, the most recently active ERVs are members of the HERV-K (HML2) family. This family first integrated into the genome of the common ancestor of humans and Old World monkeys at least 30 million years ago, and it contains >12 elements that have integrated since the divergence of humans and chimpanzees, as well as at least two that are polymorphic among humans…This recent activity makes this family ideal for distinguishing between the alternative mechanisms of proliferation. [33]
The pattern of HERV-K elements, shown below, demonstrates just how powerful the ERV evidence is in demonstrating human-ape common ancestry, as well as confirming the standard evolutionary history of primates. Again, ERVs are remnants of ancient viral infection. They are not native to humans or primates, but bear witness to ancient retroviral infection. The odds of multiple identical HERV elements integrating into primate and human DNA in exactly the right places to simulate common descent are so low as to be effectively zero.

Approximate integration times of HERV-K elements. Arrows indicate the lineage in which a particular LTR was first detected, and numbers refer to the cluster as identified in Fig. 1 Time estimates for divergence of the different primate lineages were taken from Bailey et al. From Patrik Medstrand and Dixie L. Mager Human-Specific Integrations of the HERV-K Endogenous Retrovirus Family J Virol. 1998 December; 72(12): 9782–9787.
Shared NHEJ
If DNA repair via NHEJ occurs in the germ line, then that scar of DNA repair will be inherited. The presence of identical DNA repair scars at exactly the same place in a family of human beings would be considered a good mark of consanguinity. If we see this same NHEJ repair at the same place in humans and chimps, then we'd have powerful evidence of human-chimp common ancestry. As it turns out, we don't have just one example of shared NHEJ scars in humans and chimps. We have thirty-seven. Furthermore, we have many more shared by humans, chimps and orang-utans, as well as over 300 shared by humans, apes, and monkeys, all consistent with the predictions of common descent. Cancer researcher and cell biologist Graeme Finlay notes:
Hundreds of such DNA scars have been identified. Some are shared by humans, chimps and orang-utans, but no other species (142 in total). These scars arose in a great ape ancestor. Others are shared by apes and [Old World Monkeys]. but by no other species (219). Each of these dates from an ape-OWN ancestor. Many are common to all anthropoid primates (389)…The distribution of shared repair patches generates a phylogenetic tree that is congruent with that generated from ERV and retrotransposon insertions. NHEJ and retrotransposition are wholly independent processes, and so provide wholly independent signatures that illuminate our evolutionary past. [34]
 |
| Scars representing DNA repair by NHEJ. AN example is shown (upper diagram). The length of DNA copied from elsewhere is indicated by white lettering starting ATCTT…on a dark grey background. Evolutionary staged at which NHEJ arose (lower diagram). Numerals indicate the number of repair sites identified. Source: Finlay (2013) |
Shared NUMTS
Similarly, the presence of identical mtDNA elements at the same place in the genomes of humans and apes (either as repair elements or as insertions) would be powerful evidence of common ancestry of humans and apes, with such a mtDNA insertion taking place in a common ancestor, and being subsequently inherited by both descendant lines. Shared NUMTS have been found in:
- Human and neanderthal DNA
- Humans and apes
- Humans and Old World Monkeys
- Humans, Old World Monkeys and (likely) New World Monkeys [34-38]
Finlay notes that we share most of our NUMTS with chimpanzees, with 502 of the 616 NUMTS in our genome also found in the chimpanzee genome. In fact most of the NUMTS jn our genome are also shared with old world monkeys:
The presence of individual numts in both human and chimp genomes has abundantly demonstrated he monophylicity of humans and chimps. We and the chimps are indeed sister species, derived from common ancestors. But analysis of the presence and absence of individual numts in multiple primate genomes has also been initiated. Several partial studies have shown that humans, chimps and gorillas are monophyletyic. So are the great apes (incorporating the orang-utans) and, further back in time, the apes (incorporating the lesser apes or gibbons. It appears that the majority of numts in our genome are shared with OWMs. [39]
When faced with this overwhelming evidence, science denialists have resorted to claiming that because some of these elements have been secondarily co-opted by the genome for another purpose, they are not evidence for common descent. This is a feeble explanation which ignores the fact that functionality is not the issue, but rather the presence of shared identical genomic elements which are either alien to the original site, or are performing a completely unrelated function. Finlay skewers this argument perfectly:
Are numts functional today? Quite possibly - but functionality is irrelevant to the issue of whether numts constitute markers of descent. It is the complex molecular pathway by which numts arose that makes them such compelling signatures of our shared ancestry with other primates. Each numt is a potent demonstration that the individuals and species possessing it are clonally related products of the reproductive cell in which the initiating repair event occurred. [40]A linguistic analogy makes this example clear. The presence of the same semitic loanword in all Romance languages would be best explained by the ingress of this loanword into the common ancestor of all Romance languages (Vulgar Latin), which was then subsequently inherited by the descendant languages. The fact that the loanword is still used is irrelevant to the argument of common descent.
Shared Telomeric DNA
The best-known example is the telomeric DNA in human chromosome 2 which confirms that it was the product of an ancestral fusion of two chromosomes homologous to ape chromosomes. Our understanding of the origin of chromosome 2 in a fusion event is not new. We've known about the close similarity between human and ape chromosomes as long as thirty years ago, with the researchers behind the seminal paper in Science in which this data was published noting:
"The telomeric fusion of chromosomes 2p and 2q accounts for the reduction of the 24 pairs of chromosomes of the great apes to 23 in modern man. [41]"As one can see below, the similarities are striking:

Over the following 30 years, further work has pinpointed the exact location in human chromosome 2 where the fusion event occurred. Twenty years ago, careful examination of human chromosome 2 showed that it was the result of an ancient telomere to telomere fusion:
“The inverted arrangement of the 1TAGGG array and the adjacent sequences, which are similar to sequences found at present-day human telomeres, is precisely that predicted for a head-to-head telomeric fusion of two chromosomes...These data provide strong evidence that the inverted repeats in c8.1 arose from the head-to-head fusion of ancestral telomeres.” [42]
One year later, further research showed evidence [43] of an ancient centromere in human chromosome 2, giving us evidence of both centromeric and telomeric remnant DNA which is what one would expect if human chromosome 2 was the product of a fusion event. This is no longer controversial in molecular biology. For example, a decade ago, researchers investigating the structure and evolution of human chromosome two noted in passing:
"Humans have 46 chromosomes, whereas chimpanzee, gorilla, and orangutan have 48. This major karyotypic difference was caused by the fusion of two ancestral chromosomes to form human chromosome 2 and subsequent inactivation of one of the two original centromeres (Yunis and Prakash 1982). As a result of this fusion, sequences that once resided near the ends of the ancestral chromosomes are now located in the middle of chromosome 2, near the borders of bands 2q13 and 2q14.1. For brevity, we refer henceforth to the region surrounding the fusion as 2qFus. Two head-to-head arrays of degenerate telomere repeats are found at this site; their head-to-head orientation indicates that chromosome 2 resulted from a telomere to telomere fusion. (Emphasis mine). [44]
That chromosome 2 owes its origin to a fusion event is no longer in doubt. Recently, DNA from a hominid bone found in the Denisova cave in Siberia was sequenced, showing that it came from a species closely related to the Neanderhals. It turns out that the Denisovan hominids also had the fused second chromosome. Palaeoanthropologist John Hawks quotes the researchers, noting that:
Sometime in our evolution, two separate chromosomes fused into one, giving us a karyotype of 46 chromosomes where chimpanzees, bonobos and gorillas have 48chromosomes. The high-coverage genome was sufficient to show that Denisova shared the human fusion:"Of more relevance may be examination of aspects of the Denisovan karyotype. The great apes have 24 pairs of chromosomes while humans have 23. This difference is caused by a fusion of two acrocentric chromosomes that formed the metacentric human chromosome 2 , and resulted in the unique head-to-head joining of the telomeric hexameric repeat GGGGTT. A difference in karyotype would likely have reduced the fertility of any offspring of Denisovans and modern humans. We searched all DNA fragments sequenced from the Denisovan individual and identified twelve fragments containing joined repeats. By contrast, reads from several chimpanzees and bonobos failed to yield any such fragments. We conclude that Denisovans and modern humans (and presumably Neandertals) shared a karyotype consisting of 46 chromosomes."
We still have no idea whether this fusion made any difference to any phenotype in ancient humans.
Many, many people have written me over the years to ask whether this fusion of two ancestral chromosomes might have been important to our evolution. Perhaps, many suggested, if Neandertals had a chromosomal incompatibility with us, that would explain why they became extinct. I have always doubted this, but without information it was impossible to be certain.
It's nice to now have the information in hand: This fusion happened earlier in our evolution. [45]
This poses a huge problem for special creationists. Why would God not only create humans with chromosome 2 looking like the product of a fusion of two ape chromosomes, but do the same thing to an extinct species of humans who lived thouisands of years before Adam (assuming we place Adam around the time animals and plants were domesticated in the ANE)? Common descent solves this problem neatly - the Denisovans and Homo sapiens share a common ancestor in which he chromosomal fusion took place.
More evidence for human-ape common ancestry comes from shared telomeric DNA insertion (remember, telomeric DNA belongs at the end of chromosomes. Its presence elsewhere is proof either of an insertion event or a chromosomal fusion). Finlay once again notes:
Some 50 short, well-conserved interstitial telomeric repeats…are present in the human genome. Sequencing studies indicated that they arose as distinct insertion events, probably generated by the action of the enzyme telomerase, which has the function of adding TTAGG units at the authentic telomeres. These inserts have the characteristics of emergency DNA repair patches that were recruited to hold double-stranded breaks together.The presence of absence of ten of these telomeric repeats was ascertained in humans and other primate species. ,Each repeated was shown to have arisen at a particular state of primate evolution. In some cases they precisely interrupt the pre-insertion sequence. The inserts shown…is not associated with any gains or losses of base sequence. It is a clean insertion, shared by humans, chimps and gorillas The Asian great ape (orang-utan) and OWMs retain the uninterrupted pre-insertion site. This repeat unambiguously demonstrates African great ape monophylicity. [46]
 |
| Interstitial telomeric repeats shared by the african great apes (upper diagram) and the apes (lower diagram). Source: Finlay (2013) |
Special creationist errors in rebutting this evidence
THis evidence is overwhelming, compelling, and has been accepted by > 99% of competent, professional biologists as confirming the reality of common descent. Burges' attempt to rebut this evidence is frankly pitiful. Burges asserts:
The leader of [the human genome] project, Francis Collins, a leading proponent of theistic evolution, has pointed out that substantial stretches of DNA in the genomes are very similar and so imply a common ancestor. For instance, the human and mouse genomes are roughly the same size, and the inventory of protein-coding genes is remarkably similar. Collins acknowledges that this could be the result of the Creator using successful design principles over and over again, yet states there is no current evidence from molecular biology that they need to be so similar. [47]
Burges however carefully avoids directly quoting Collins, and when we see what Collins actually says, and go past the end of Burges' indirect quote of Collins, we can see that Burges has shamefully misrepresented Collins:
The study of genomes leads inexorably to the conclusion that we humans share a common ancestor with other living things. Some of that evidence is shown in Table 5.1, where the similarity between the genomes of ourselves and other organisms is displayed. This evidence alone does not, of course, prove a common ancestor; from a creationist perspective, such similarities could simply demonstrate that God used successful design principles over and over again. As we shall see, however, and as was foreshadowed above by the discussion of "silent" mutations in protein-coding regions, the detailed study of genomes has rendered that interpretation virtually untenable— not only about all other living things, but also about ourselves. (Emphasis mine) [48]
Either Burges has simply parroted what someone has told him without bothering to verify the reference, a blunder which demonstrates appalling research skills, or he has deliberately misrepresented Collins. If we continue, we can see that Collins is even more emphatic that the evidence overwhelmingly favours common descent:
Even more compelling evidence for a common ancestor comes from the study of what are known as ancient repetitive elements (AREs). These arise from "jumping genes," which are capable of copying and inserting themselves in various other locations in the genome, usually without any functional consequences. Mammalian genomes are littered with such AREs, with roughly 45 percent of the human genome made up of such genetic flotsam and jetsam. When one aligns sections of the human and mouse genomes, anchored by the appearance of gene counterparts that occur in the same order, one can usually also identify AREs in approximately the same location in these two genomes.
Some of these may have been lost in one species or the other, but many of them remain in a position that is most consistent with their having arrived in the genome of a common mammalian ancestor, and having been carried along ever since. Of course, some might argue that these are actually functional elements placed there by the Creator for a good reason, and our discounting of them as "junk DNA" just betrays our current level of ignorance. And indeed, some small fraction of them may play important regulatory roles. But certain examples severely strain the credulity of that explanation.
The process of transposition often damages the jumping gene. There are AREs throughout the human and mouse genomes that were truncated when they landed, removing any possibility of their functioning. In many instances, one can identify a decapitated and utterly defunct ARE in parallel positions in the human and the mouse genome .
Unless one is willing to take the position that God has placed these decapitated AREs in these precise positions to confuse and mislead us, the conclusion of a common ancestor for humans and mice is virtually inescapable. This kind of recent genome data thus presents an overwhelming challenge to those who hold to the idea that all species were created ex nihilo. (Emphasis mine) [49]
As Collins notes, many of these retrotransposons are dead on arrival - they have no possibility of being co-opted by evolution to serve another function; the creationist attempt to explain why nearly 45% of our genome consists of these mobile genetic parasites by asserting that they are all functional severely strains credibility, to paraphrase Collins.
Special creationists such as Burges simply have no way of explaining the multiple shared identical genomic 'errors' at identical places in human and ape genomes, so their approach, as Collins notes, is to suddenly change the question, and assert - based on a tiny example of pseudogenes or retrotransposons that have been co-opted to serve a completely different function - that they are all functional. As Finlay says, the functionality of these elements is not the issue. The issue is genetic plagiarism - the presence of identical genetic 'mistakes' at the same place in multiple genomes, and Burges simply ignores it. He continues by asserting:
The argument continues that the genomes contain long sections of DNA that do not code for proteins and are littered with 'ancient repetitive elements', referred to as genetic 'flotsam and jetsam', 'pseudogenes', or 'junk DNA;, with no apparent function. When these turn up at the same places in the DNA of different species, it is claimed that these species must share a common ancestry. The recent international ENCODE project, however, has shown that in fact more than eighty percent of the human genomes' components are biologically active, and that most of these non-coding sections of our genome are not junk at all but perform vital functions in our cells. [50]
Burges clearly has not read or understood Collins who flatly asserts that the presence of multiple non-functional retrotransposons in human and mice DNA "presents an overwhelming challenge to those who hold to the idea that all species were created ex nihilo." As we've seen earlier, the evidence from shared identical DNA repair scars alone confirms human-ape common ancestry, let alone the data from nuclear DNA, retrotransposon insertions, ERV insertions, pseudogenes, NUMTs, NHEJs, and telomeric insertions. Burges provides no substantive rebuttal, but instead changes the subject, and harps on the alleged functionality of junk DNA. Once again, functionality is not the issue. Rather, it is the presence of shared identical genetic 'errors' at the same place. Burges simply ignores this, and hopes that his readers will not notice his clumsy bait and switch approach.
ENCODE - The truth is that at least 66% of our DNA is worthless junk
Anyone who appeals to the ENCODE data in an attempt to rebut the evidence for common descent is merely broadcasting their ignorance of the fact that the ENCODE team and results have been heavily criticised by many evolutionary biologists. For those unaware of what ENCODE is, some context will be provided.
In 2001, the human genome was sequenced [51]. Over the past nine years, the Encyclopedia of DNA elements (ENCODE) project has been examining the genome in order to examine what the genome does. Now, the ENCODE project has released several papers announcing the results of its research. One of results of its research is that "more than 80% of the human genome's components have now been assigned at least one biochemical function." [52] How does this square with the fact that much of our genome is made up of non-coding DNA such as retrotransposons (nearly half the DNA), intronic DNA or endogenous retroviral elements?
The key word here is 'functional'. I have to stress that functional does not mean essential or beneficial. Retrotransposons for example have been linked with human disease. [53] We would be arguably better off if those SINEs were silent. Special creation already has to accept that if every nucleotide was created by God, then the creator has deliberately inserted genomic material which causes immense misery in the human race. The intelligent designer becomes a malevolent designer if the logic of the special creationist position is carried through to its inevitable conclusion.
Another point to remember is that being transcribed counts as biological function, irrespective of whether that transcribed section actually does something beneficial for the organism. Without this context, claims that the 80% figure invalidate what we already know about the genome (that most of it is non-coding junk) can be dismissed.
There is of course no substitute for informed commentary (as opposed to special creationist disinformation), which is why the opinions of senior scientists involved in the ENCODE project are worth reading. Ewan Birney, the lead analysis coordinator for ENCODE over the past five years is arguably a man whose opinion would count for something. So what does he say about the 80% figure:
It’s clear that 80% of the genome has a specific biochemical activity – whatever that might be. This question hinges on the word “functional” so let’s try to tackle this first. Like many English language words, “functional” is a very useful but context-dependent word. Does a “functional element” in the genome mean something that changes a biochemical property of the cell (i.e., if the sequence was not here, the biochemistry would be different) or is it something that changes a phenotypically observable trait that affects the whole organism? At their limits (considering all the biochemical activities being a phenotype), these two definitions merge. Having spent a long time thinking about and discussing this, not a single definition of “functional” works for all conversations. We have to be precise about the context. Pragmatically, in ENCODE we define our criteria as “specific biochemical activity” – for example, an assay that identifies a series of bases. This is not the entire genome (so, for example, things like “having a phosphodiester bond” would not qualify). We then subset this into different classes of assay; in decreasing order of coverage these are: RNA, “broad” histone modifications, “narrow” histone modifications, DNaseI hypersensitive sites, Transcription Factor ChIP-seq peaks, DNaseI Footprints, Transcription Factor bound motifs, and finally Exons.(Emphasis mine) [54]
Specific biochemical activity does not mean essential to life. This is the point that Burges completely ignores.This point needs to be hammered home to every special creationist who latches onto the ENCODE paper and claims that 80% of the genome is functional (though one wonders why they are still happy to accept the implication that God created the human genome with 20% junk). Birney continued by commenting on with what definition of 'functional he is happy:
Back to that word “functional”: There is no easy answer to this. In ENCODE we present this hierarchy of assays with cumulative coverage percentages, ending up with 80%. As I’ve pointed out in presentations, you shouldn’t be surprised by the 80% figure. After all, 60% of the genome with the new detailed manually reviewed (GenCode) annotation is either exonic or intronic, and a number of our assays (such as PolyA- RNA, and H3K36me3/H3K79me2) are expected to mark all active transcription. So seeing an additional 20% over this expected 60% is not so surprising.
However, on the other end of the scale – using very strict, classical definitions of “functional” like bound motifs and DNaseI footprints; places where we are very confident that there is a specific DNA:protein contact, such as a transcription factor binding site to the actual bases – we see a cumulative occupation of 8% of the genome. With the exons (which most people would always classify as “functional” by intuition) that number goes up to 9%. Given what most people thought earlier this decade, that the regulatory elements might account for perhaps a similar amount of bases as exons, this is surprisingly high for many people – certainly it was to me!
In addition, in this phase of ENCODE we did sample broadly but nowhere near completely in terms of cell types or transcription factors. We estimated how well we have sampled, and our most generous view of our sampling is that we’ve seen around 50% of the elements. There are lots of reasons to think we have sampled less than this (e.g., the inability to sample developmental cell types; classes of transcription factors which we have not seen). A conservative estimate of our expected coverage of exons + specific DNA:protein contacts gives us 18%, easily further justified (given our sampling) to 20% (Emphasis mine)
In other words, once we start changing our definition of 'functional' to one more consistent with what the layperson would take it to be (ie: biologically useful or essential) the 80% figure drops to around 20%. As for why ENCODE emphasised the 80% figure, rather than the 20% one more consistent with that the layperson would perceive 'functional' to mean, Birney states:
Originally I pushed for using an “80% overall” figure and a “20% conservative floor” figure, since the 20% was extrapolated from the sampling. But putting two percentage-based numbers in the same breath/paragraph is asking a lot of your listener/reader – they need to understand why there is such a big difference between the two numbers, and that takes perhaps more explaining than most people have the patience for. We had to decide on a percentage, because that is easier to visualize, and we choose 80% because (a) it is inclusive of all the ENCODE experiments (and we did not want to leave any of the sub-projects out) and (b) 80% best coveys the difference between a genome made mostly of dead wood and one that is alive with activity. (Emphasis mine)
Alive with activity again does not mean essential to life. A retrotransposon that copies and pastes itself indiscriminately in the genome is functional, but when it causes genetic disorders it is clearly not beneficial. Unsurprisingly, special creationists tend to ignore the 45% of the genome that is retrotransposed DNA, essentially parasitic genetic material.
The ENCODE hype has been criticised severely for its misleading 80% figure. Dan Graur et al have published a takedown of the extravagant ENCODE claims:
The ENCODE hype has been criticised severely for its misleading 80% figure. Dan Graur et al have published a takedown of the extravagant ENCODE claims:
It is worth noting that when Ewan Birney, the lead scientist for ENCODE was pressed on the claim that 80% of the genome is essential to life, he conceded that this was not true. In a BBC Radio interview, Birney admitted:A recent slew of ENCODE Consortium publications, specifically the article signed by all Consortium members, put forward the idea that more than 80% of the human genome is functional. This claim flies in the face of current estimates according to which the fraction of the genome that is evolutionarily conserved through purifying selection is under 10%. Thus, according to the ENCODE Consortium, a biological function can be maintained indefinitely without selection, which implies that at least 80 – 10 = 70% of the genome is perfectly invulnerable to deleterious mutations, either because no mutation can ever occur in these “functional” regions, or because no mutation in these regions can ever be deleterious. This absurd conclusion was reached through various means, chiefly (1) by employing the seldom used “causal role” definition of biological function and then applying it inconsistently to different biochemical properties, (2) by committing a logical fallacy known as “affirming the consequent,” (3) by failing to appreciate the crucial difference between “junk DNA” and “garbage DNA,” (4) by using analytical methods that yield biased errors and inflate estimates of functionality, (5) by favoring statistical sensitivity over specificity, and (6) by emphasizing statistical significance rather than the magnitude of the effect. Here, we detail the many logical and methodological transgressions involved in assigning functionality to almost every nucleotide in the human genome. The ENCODE results were predicted by one of its authors to necessitate the rewriting of textbooks. We agree, many textbooks dealing with marketing, mass-media hype, and public relations may well have to be rewritten. (Emphasis mine) [55]
Chris Ponting: So I think we can probably agree between us that between 10% and say 20% is vital for life.Ewan Birney: I mean, I think we would agree with that. I think, you know, refining that percentage down is quite interesting. I think also the other components that we — biochemical events that we see in the genome, sort of, each one of them are equally likely to be part of that 10% to 20% that we’re looking for. It’s important to realize that it’s not the case that we can spot the 10% to 20% just by looking harder. Each of these different places in the genome that have some biochemical activity associated with it, when there’s some phenotype screen that’s directed there or some evolutionary screen that’s directed to that point, ENCODE can now say “Ah ha! Here is a biochemical thing that this piece of DNA looks like it could be doing”. (Emphasis mine) [56]
Evolutionary biologist TR Gregory who is also an expert in genome size evolution - putting him in a perfect position to provide informed commentary on the subject has taken a considerable interest in the subject. In the comments section of one of Gregory's blog posts discussing the ENCODE hype, respected evolutionary geneticist Joe Felsenstein makes a penetrating comment which cuts to the heart of the hype:
Ewan Birney is trying to give the impression that the problem is that people have misinterpreted him. But he was the one who put forward the 80% figure. It was not added by the popular science press, he wanted it out there and wanted it noticed. And when there was a huge blaze of publicity centered on the (purported) death of junk DNA, publicity that Ryan has done us the great service of listing, I didn’t notice Birney jumping up saying that he had been misinterpreted.
Birney later admitted on bis bog that the 80% figure represented biological activity, which was not the same thing as essential to life.Large numbers of laypeople and other scientists are now persuaded that there never was any junk DNA. It will probably take 10 years to unpersuade them. We have Birney to thank for this situation. I’m saddened to see him dance around and try to give the impression that someone else came up with the Death of Junk DNA. (Emphasis in the original) [57]
The problem with 'science by press release', is that in order to gain the attention of your audience, there is a very real temptation to succumb to hyperbole, and when you are dealing with the general public, terms such as 'functional' need to be defined properly, otherwise there is the chance that they will get the wrong idea. Certainly, when most people hear 'functional', they are likely to think that 80% of the genome is essential to life, which is simply false. As project leader Ewan Birney acknowledged later, the 80% figure represents biological activity, which is definitely not the same thing as functional:
Q. Ok, fair enough. But are you most comfortable with the 10% to 20% figure for the hard-core functional bases? Why emphasize the 80% figure in the abstract and press release?
A. (Sigh.) Indeed. Originally I pushed for using an “80% overall” figure and a “20% conservative floor” figure, since the 20% was extrapolated from the sampling. But putting two percentage-based numbers in the same breath/paragraph is asking a lot of your listener/reader – they need to understand why there is such a big difference between the two numbers, and that takes perhaps more explaining than most people have the patience for. We had to decide on a percentage, because that is easier to visualize, and we choose 80% because (a) it is inclusive of all the ENCODE experiments (and we did not want to leave any of the sub-projects out) and (b) 80% best coveys the difference between a genome made mostly of dead wood and one that is alive with activity. We refer also to “4 million switches”, and that represents the bound motifs and footprints.
We use the bigger number because it brings home the impact of this work to a much wider audience. But we are in fact using an accurate, well-defined figure when we say that 80% of the genome has specific biological activity. [58]
In other words, between 10-20% of the genome consists of 'hard core functional bases' with the rest simply being biologically active, which is not the same thing as essential to life. Retrotransposable elements that copy and paste themselves randomly into the genome are biologically active, but hardly essential - or beneficial, as evidenced by the genetic diseases connected to retrotransposable DNA. Even if one grants that the entire 80% figure refers to essential genomic material, that still leaves 20% of the genome non-coding, non-functional junk, which is inconsistent with the idea that the genome is the product of an intelligent designer.
Since then, the much-touted 80% figure is changing. Science journalist Faye Flam contacted John Stamatoyannopoulos, one of the ENCODE researchers to clarify the 80% figure. It turns out that it is more like 40%:
He said he thought the skeptics hadn’t fully understood the papers, and that some of the activity measured in their tests does involve human genes and contributes something to our human physiology. He did admit that the press conference mislead people by claiming that 80% of our genome was essential and useful. He puts that number at 40%. Otherwise he stands by all the ENCODE claims. (Emphasis mine) [59]
So, we can safely bin the "80% of the genome is functional" claim as even researchers from ENCODE are backing away from it.
Max Libbrecht, another ENCODE researcher also commented on the ENCODE debacle, showing that even members of the project realised just how damaging the "80% is functional" hype was:
After I took part in an AMA ("Ask Me Anything") on reddit, there has been some discussion elsewhere (such as by Ryan Gregory and in the comments of Ewan Birney's blog) of what I and the other ENCODE scientists meant. In response, I'd like to echo what many others have said regarding the significance of ENCODE on the fraction of the genome that is "junk" (or nonfunctional, or unimportant to phenotype, or evolutionarily unconserved).
In its press releases, ENCODE reported finding 80% of the genome with "specific biochemical activity", which turned into (through some combination of poor presentation on the part of ENCODE and poor interpretation on the part of the media) reports that 80% of the genome is functional. This claim is unlikely given what we know about the genome (here is a good explanation of why), so this created some amount of controversy.I think very few members of ENCODE believe that the consortium proved that 80% of the genome is functional; no one claimed as much on the reddit AMA, and Ewan Birney has made it clear on his blog that he would not make this claim either. In fact, I think importance of ENCODE's results on the question of what fraction of DNA is functional is very small, and that question is much better answered with other analysis, like that of evolutionary conservation. Lacking proof either way from ENCODE, there was some disagreement on the AMA regarding what the most likely true fraction is, but I think this stemmed from disagreements about definitions and willingness to hypothesize about undiscovered function, not misinterpretation of the significance of ENCODE's results.
I think many members of the consortium (including Ewan Birney) regret the choice of terminology that led to the misinterpretations of the 80% number. Unfortunately, such misinterpretations are always a danger in scientific communication (both among the scientific community and to the public). Whether the consortium could have done a better job explaining the results, and whether we should expect the media to more accurately represent scientific results, is hard to say.
I think the contribution of ENCODE lies not in determining what DNA is functional but rather in determining what the functional DNA actually does. This was the focus of the integration paper and the companion papers, and I would have preferred for this to be the focus of the media coverage. (Emphasis mine) [60]
In short:
- The claim that 80% of the genome is essential to life is false. The figure is more like 10-20%
- The value of ENCODE, to quote one of its researchers is in determining what the functional DNA actually does, rather than how much is functional.
- The question of functionality does not take away the considerable evidence for common descent. Burges has completely failed to address in any substantive way this evidence, and the ENCODE diversion merely demonstrated his ignorance of the controversy surrounding ENCODE and the acknowledgement that the 80% figure was hype.
How much of the genome is actually essential to life? Not much. Around 45% of the genome is made up of mobile genetic elements - retrotransposons - that copy and paste themselves into the genome randomly, often causing disease in the process. This is very much an unguided, random process. A significant fraction of the human genome owes its origin to ancient retroviral infection. In fact, there is more retroviral genetic material – the evidence of past retroviral infection – in our genome than there is direct protein coding material. Only a a small percentage of the human genome directly codes for protein or has specific regulatory function.
Breaking down the human genome into the various classes of genetic material we find there, the scale of how much parasitic DNA, decayed viral remnants and genetic equivalent of gibberish [60] is astonishing:
Transposable Elements: 44% junk
DNA transposons: functional < 0.1%, defective 3%
Retrotransposons: active < 0.1%, co-opted < 0.1%, junk 41%
Viruses: 9% junk
DNA Viruses: active < 0.1%, defective ~1%
RNA Viruses: active < 0.1%, co-opted < 0.1%, defective 8%
Pseudogenes: 1.2% junk
Derived from protein-coding genes: 1.2% junk
Co-opted pseudogenes: < 0.1% useful, secondarily acquired new function
Ribosomal RNA genes: 0.19% junk
Essential: 0.22%
Junk: 0.19%
Other RNA encoding genes
tRNA genes: < 0.1% essential
known small RNA genes: < 0.1% essential
putative regulatory genes: ~2% essential
Protein-encoding genes: 9.6% junk (intron sequences), 1.8% essential transcribed
Regulatory Sequences: 0.6% essential
Origins of DNA replication: < 0.1% essential
Scaffold attachment regions: < 0.1% essential
Highly repetitive regions: 1% junk, 2% essential
Intergenic DNA: 26.3% unknown function, most likely junk, 2% essential
Essential / Functional DNA: 8.7%
Junk DNA: 65%
Unknown: 26.3%
Even if most of the intergenic DNA turns out to have a function, nearly 66% of our genome is rubbish consisting of remnants of ancient retroviral infection, damaged genes that can no longer work, mobile genetic elements that copy and insert themselves randomly around the genome irrespective of what benefit or harm that action does, and introns, the non-coding sections of DNA that interrupt genes.
When in doubt, appeal to ignorance and imply those who accept evolution are backsliders
Burges in a marginal reference makes a desperate attempt to explain away the genomic evidence, which starta with an appeal to ignorance - maybe the retroviral elements will turn out to have a function after all - and degenerates to the point where he ends up making wild, utterly unprovable assertions that the Fall somehow warped the genomes of humans and animals. I am not making this up, sadly:
For instance, the human genome contains sequences of what appear to be inactive retrovirus genomes incorporated in the same specific places in the genomes of everyone. The hypothesis is that these are the results of random insertions into the genome of a common human ancestor, since the same apparent 'ancient' retrovirus sequences are located in exactly the same equivalent place, between the same genes, in the equivalent chromosomes of some primates, including bonobos, chimps, and gorillas (although not in those of others such as orang-utans). [62]

LTR—gag—pol—env—LTR
LTR: long terminal repeat. They are DNA sequences that are involved in the insertion of the retroviral genome into host DNA.
gag: group specific antigen. This codes for retroviral structural proteins
pol: polymerase. This codes for reverse transcriptase, protease and integrase
env: envelope. This codes for the retroviral coat proteins.
It is hard not to miss the decayed remnants of retroviral infection when we see the decayed remains of retroviruses littering the genome. Furthermore, we know that these ERVS are alien to the vertebrate genome because of codon bias. As ERV researcher Abigail Smith notes:
Viruses, including retroviruses, including endogenous retroviruses, dont speak the same language as humans. Sure they use A, T/U, C, G nucleotides in codons, coding for amino acids that make proteins.
But viruses and humans dont speak this language with the same accent. Its called codon bias, or codon-pair bias.
Several codons can code for the same amino acid. For instance, GCU, GCC, GCA, GCG all code for Alanine. So, in the human genome, you would expect ~25% of all the Alanines to use GCU, ~25% to use GCC, ~25% to use GCA, and ~25% to use GCG, right? Nope. For some reason, the human genome prefers GCC over GCG. Four times as many Alanines are coded by GCC as GCG. Humans have a GCC ‘accent’.
Viruses have their own codon ‘accents’ as well. And even though the differences should, theoretically, mean nothing (an Alanine is an Alanine is an Alanine), one way we can attenuate viruses to make better vaccines is to force them to use codons they dont like.
Thus one of the ways we know ERVs are a later addition to the human genome, and didnt originate in the human genome, is that retroviruses have a different codon ‘accent’ than humans. They don't fit. [63]
Furthermore, we have evidence of retroviral insertion in real-time:
Since ERVs have been found in every vertebrate whose genome has been examined, it is not unreasonable to look for both evidence of active ERV infection and examples of ERV inclusions in other related species. This is in fact what we see. Take the subject of active retroviral infection. The koala genome is currently being colonised by the koala retrovirus (KoRV). Research [64] has shown that:
KoRV is present, at variable copy number, in the germline of all koalas found in Queensland, but that animals from some areas of southern Australia lack the provirus. Most notably, KoRV appears completely absent from koalas on Kangaroo Island off the coast of South Australia. This island was stocked with koalas in the early part of the twentieth century and has remained essentially isolated since then; it appears most likely that the small founding population was entirely free of KoRV. Tarlington et al. suggest that an ongoing process of infection and endogenization is now occurring, spreading from a focus in northern Australia that quite possibly initiated within the last 100 to 200 years. [65]
This by the way is not something that is of particular immediate benefit to the koala:
KoRV appears to be associated with the fatal lymphomas that kill many captive animals . It may also be immunosuppressive, thereby contributing to the chlamydial infections that afflict many koalas . [66]
There is no doubt that what we see in our genome is the result of ancient retroviral infection, and as I pointed out earlier, respected virologist John Coffin notes:
As a rule, they entered the germ line before the origin of the species, and are therefore found in every individual at the same genomic location. Because of their long residence in the host DNA, they have accumulated mutations and other genetic damage that cause them to be inactive, and, in general, they cannot be activated to yield infectious virus. Because the site of integration in the genome, which comprises some three billion base pairs in humans, is essentially random, the presence of an ancient provirus at exactly the same position in different, but related, species cannot occur by chance, but must be a consequence of integration into the DNA of a common ancestor of all the species that contain it. [67]
Therefore, when Burges continues by asserting:
Time may tell whether these are actually ancient retroviral sequences or whether they have some other function.
he is completely and utterly wrong. These are remains of ancient retroviral sequences. Human and primate genomes are littered with the remains of ancient retroviral infection, and as he motes, the presence of an ancient proviral element at the same place in different but related species is evidence of common ancestry.
Furthermore, Burges, like almost all special creationist critics of evolution, has confused ERV elements - such as LTRs and the retroviral genes gag, pol, and env - with a fully functional ERV. Occasionally, evolution co-opts ERV elements but this is not the same thing as a fully functional ERV which would simply produce more retroviruses. One does not want a fully functional ERV in the genome producing retroviruses, given the disease-causing potential of retroviruses such as HIV. Burges' complete ignorance of retrovirology and genomics is painfully apparent at this point.
Things get worse when he blithely asserts:
The argument that common gene sequences must mean common ancestry (as opposed to common design) is an assertion which disregards alternative explanations. Remember, that the argument starts fro, the premise that all primates descended from common ancestors.
Burges ignores the fact that the common design alternative has been examined and discarded. There is here a subtle shift from shared identical proviral elements to common gene sequences, a rhetorical shift which frankly is evasive. Even then, this does not help Burges. As I've mentioned before, the considerable functional redundancy in the genetic code means that for even a small 100 amino acid protein, there are something like 10^49 possible ways to code for exactly the same protein. Common descent would predict that the gene sequences for all life today would be clustered together in such a way that the phylogenetic tree constructed from the genomic data would be consonant with the standard phylogenetic tree. Conversely, under a model of special creation, there would be no reason for humans and chimps to have gene sequences closely related. However, this is exactly what we see. The special creation hypothesis is therefore rejected.
Getting back to the question of shared identical genetic 'mistakes' at the same place, as Francis Collins pointed out:
Unless one is willing to take the position that God has placed these decapitated AREs in these precise positions to confuse and mislead us, the conclusion of a common ancestor for humans and mice is virtually inescapable. This kind of recent genome data thus presents an overwhelming challenge to those who hold to the idea that all species were created ex nihilo.
The common design explanation is as Collins said, virtually inescapable.
Conclusion
Alas, the only possible escape route is the one to which Burges desperately appeals, and that is the 'God buried the fossils to deceive mankind' argument that some fundamentalists use. Burges in all seriousness appeals to this argument in a desperate attempt to salvage his special creationism:
It is sometimes argued that these features of DNA look like common descent, and therefore must evidence it, because otherwise God would be deceiving us. But while He is certainly "the God of truth", it is also true that, for those who have turned from Him, He can send strong delusion that they should believe a lie. It is those who absolutely trust His Word who will not be deceived." [68]
This is ignorant and grossly offensive on a scale that I have scarcely seen before in Christadelphian material. It is an outrageous slur on fellow believers who unlike Burges have the intellectual honesty to follow the evidence where it leads, irrespective of what human interpretations of the Bible assert, to claim that they have 'turned from Him.' That he has resorted to these tactics confirms just how intellectually threadbare his antievolution polemic is.
This is confirmed by his shambolic exegesis of 2 Thess 2:11 to imply that God has deliberately created the illusion of common descent, down to faked ERV inclusions, fictitious examples of DNA repair, and a bloated genome filled with non-functioning junk DNA, in order to trick people. 2 Thess has nothing to do with modern science. Burges has simply wrested an isolated verse completely out of context in order to give a veneer of Biblical authority to his science denialism.
His final comment "It is those who absolutely trust His Word who will not be deceived" is smug, patronising, and fideistic to the core. In other words, Burges is commanding his fellow science denialists to close their eyes and ears to the witness of creation, and shout as loud as they an in order to drown out any evidence that threatens their fundamentalism. It also shows sadly how far our community has degenerated from the noble example set by its early members with respect to how it engaged with the scientific evidence. CC Walker had the honesty to admit that if scientific evidence contradicted dogma, then the only intellectually honest approach would be to change our interpretation of the Bible. In commenting on ancient giant bird fossils, he noted:
Supposing that it were ever established that they were the actual progenitors of our smaller forms (“There were giants in the earth in those days” might apply to birds and beasts), would the credibility of the Mosaic narrative suffer? Not at all, in our estimation. We should indeed have to revise somewhat our interpretation of the brief cosmogony of Gen. 1.; but should not waver as concerning its divinity, nor await with less faith and patience the reappearance of Moses in the land of the living.’ (Emphasis mine) [69]
We would do well to emulate that spirit of intellectual honesty
References
1. Burges D "Is Theistic Evolution Compatible With Faith in God's Word?" The Testimony (2014) 84:143-147
2. Behe M The Edge of Evolution: The Search for the Limits of Darwinism (2007: Free Press) p 71-72
3. Ostertag E.M. et al "SVA Elements Are Nonautonomous Retrotransposons that Cause Disease in Humans" Am J Hum Genet (2003) 73:1444-1451
4. Callinan, P. and Batzer, M.A. (2006) Retrotransposable elements and human disease. In Genome and Disease. Genome Dynamics (Vol. 1) (Volff, J., ed.), pp. 104–115, Karger
5. Crow, Mary K. "Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease." Autoimmunity (2009) 43: 7-16.
6. Schneider, Anna M., et al. "Roles of retrotransposons in benign and malignant hematologic disease." Cellscience (2009) 6:121.
7. Gui W, Zhang F, Lupski RJ "Mechanisms for human genomic rearrangements" PathoGenetics (2008) 1:4
8. Karaguchi H, O'hUign C, Klein J "Evolutionary Origin of Mutations in the Primate Cytochrome P450c2l Gene" Am. J. Hum. Genet. (1992) 50:766-780
9. ibid, p 777.
10. Wafaei J.R., and Choi F.Y., "Glucocerebrosidase Recombinant Allele: Molecular Evolution of the Glucocerebrosidase Gene and Pseudogene in Primates" Blood Cells, Molecules, and Diseases 35 (2005) 35: 277-85.
11. Haag F, Koch-Nolte F, Kiihl M, et al. Premature stop codons inactivate the RT6 genes of the human and chimpanzee species. J Mol Biol (1994) 243:537-546
12. Xiangwei Wu, Donna M. Muzny, Cheng Chi Lee, C. Thomas Caskey "Two independent mutational events in the loss of urate oxidase during hominoid evolution" Journal of Molecular Evolution (1992) 34:78-84
13. L.-Y.Edward Chang, Jerry L. Slightom "Isolation and nucleotide sequence analysis of the β-type globin pseudogene from human, gorilla and chimpanzee" Journal of Molecular Biology (1984) 180:767:783
14. W. C. Wong et al., "Comparison of Human and Chimpanzee Zeta 1 Globin Genes" Journal of Molecular Evolution (1985) 22: 309-15
15. Ohta Y, Nishikimi M. "Random nucleotide substitutions in primate nonfunctional gene for L-gulonogamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis" Biochim Biophys Acta (1999) 1472:408-11.
16. Fairbanks D.J. "Relics of Eden: The Powerful Evidence of Evolution in Human DNA" (2007, Prometheus Books) p 54
17. Ostertag E.M. et al "SVA Elements Are Nonautonomous Retrotransposons that Cause Disease in Humans" Am J Hum Genet (2003) 73:1444-1451
18. Callinan, P. and Batzer, M.A. (2006) Retrotransposable elements and human disease. In Genome and Disease. Genome Dynamics (Vol. 1) (Volff, J., ed.), pp. 104–115, Karger
19. Crow, Mary K. "Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease." Autoimmunity 43.1 (2009): 7-16.
20. Schneider, Anna M., et al. "Roles of retrotransposons in benign and malignant hematologic disease." Cellscience 6.2 (2009): 121.
21. Sawada I. et al "Evolution of alu family repeats since the divergence of human and chimpanzee" Journal of Molecular Evolution (1985) 22:316-322
22. Scott W. Herke S.W. et al "A SINE-based dichotomous key for primate identification" Gene (2007) 390:39-51
23. ibid, p 43
24. Novick G.E. et al. "Polymorphic human specific Alu insertions as markers for human identification." Electrophoresis (1995) 16: 1596-1601.
25. Coffin JM “Evolution of Retroviruses: Fossils in our DNA” Proceedings of the American Philosophical Society (2004) 148:264-280
26. Johnson WE Coffin JM Constructing primate phylogenies from ancient retrovirus sequences Proc. Natl. Acad. Sci USA (1999) 96:10254-10260
27. ibid p 10254
28. ibid p 10255
29. Johnson WE Coffin JM op cit p 10255-10256
30. ibid p 10256
31. ibid p 10259
32. Barbulescu M et al “Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans” Current Biology (1999) 9:861-868
33. Belshaw R et al “Long-term reinfection of the human genome by endogenous retroviruses” Proc. Natl. Acad. Sci. USA. (2004) 101:4894-4899
34. Finlay G Human Evolution: Genes, Genealogies and Phylogenies (2013: Cambridge University Press) p 140-141
35. Zischler H, Geisert H, Castresana J "A hominoid-specific nuclear insertion of the mitochondrial D-loop: implications for reconstructing ancestral mitochondrial sequences" Molecular Biology and Evolution (1998) 15:463-469
36. Zischler H "Nuclear integrations of mitochondrial DNA in primates; inferences of associated mutational events" Electrophoresis (2000) 21:531-536
37. Schmitz H, Ohme M, Zischler H "The complete mitochondrial sequence of Tarsius bancanus: evidence for an extensive nucleotide compositional plasticity of primate mitochondrial DNA." Molecular Biology and Evolution (2002) 19:544-554
38. Ovchinnikov IV, Kholina OI "Genome digging: insight into the mitochondrial genome of Homo." PLoS ONE (2010) 5,e14278
39. Finlay, op cit p 144-145
40. ibid, p 145
41. Yunis J.J. Prakash O, "The origin of man: a chromosomal pictorial legacy". Science (1982)215:1525–1530.
42. IJdo JW, Baldini A, Ward DC, Reeders ST, Wells RA, Origin of human chromosome 2: an ancestral telomere-telomere fusion. Proc Natl Acad Sci USA (1991) 88:9051-5
43. Avarello R et al "Evidence for an ancestral alphoid domain on the long arm of human chromosome 2" Hum Genet (1992) 89:247-9
44. Fan Y, Linardopoulou E, Friedman C, et al "Genomic Structure and Evolution of the Ancestral Chromosome Fusion Site in 2q13–2q14.1 and Paralogous Regions on Other Human Chromosomes" Genome Res. (2002) 12:1651-1662
45. Hawks, J "The fused chromosome 2 was in Denisova" John Hawks Weblog 1st Sep 2012
46. Finlay, op cit p 146-147
47 Burges, op cit p 145
48 Collins FS The Language of God: A Scientist Presents Evidence for Belief (2006: Free Press) p 133-134
49 ibid, p 135-137
50. Burges, op cit p 145-146
51. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome Nature (2001) 409:860-921
52. Skipper M, Dhand R, Campbell P "Presenting ENCODE" Nature (2012) 489:45 doi:10.1038/489045a
53. Prescott L. Deiningera PL and Batzerc MA "Alu Repeats and Human Disease" Molecular Genetics and Metabolism (1999) 67:183-193
55. Graur D et al "On the Immortality of Television Sets: “Function” in the Human Genome According to the Evolution-Free gospel of ENCODE" Genome Biol Evol (2013) 5:578:590
57. Gregory TR "BBC Interview with Ewan Birney" Genomicron April 1 2013. Comment
59. Flam F "Skeptical Takes on Elevation of Junk DNA and Other Claims from ENCODE Project" Tracker: Peer Review Within Science Journalism 12 Sep 2012
60. Libbrecht M "On ENCODE's results regarding junk DNA" mlibbrecht Oct 8 2012
62. Burges, op cit p 146
63. Smith A "Claim 8– ERVs were Specially Created portions of the human genome before The Fall" Oct 6 2009
65. Stoye JP “Koala retrovirus: a genome invasion in real time” Genome Biology 2006, 7:241-3
66. ibid, p 243
68. Burges op cit p 146
69. Walker, 'Genesis', The Christadelphian (1910) 47:501
No comments:
Post a Comment
Please do not comment as 'Anonymous'. Rather, choose 'Name/URL' and use a fake name. The URL can be left blank. This makes it easier to see who is replying to whom.